Tag: Actualité

Il faut être trois pour danser le tango : la synapse tripartite révélée par la microscopie superresolutive
Les astrocytes sont les cellules gliales les plus nombreuses dans le cerveau. Elles utilisent les signaux calciques pour réguler leur…

Intestin et cerveau dans la maladie de Parkinson : une voie à double sens
Plusieurs études ont récemment montré que la maladie de Parkinson pourrait trouver son origine dans l’intestin. En effet, des agrégats…

Expression enrichie des gènes de l’obésité dans des régions cérébrales clefs de l’addiction et de la récompense
L’obésité, qui est une maladie multifactorielle d’origine génétique et environnementale, est un enjeu majeur de santé publique dont la prévalence…
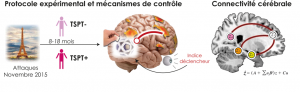
Nouvelles pistes pour comprendre la résilience au trauma
Les attentats de Paris et Saint-Denis, le 13 novembre 2015, ont laissé des marques durables, non seulement sur les survivants…

Connectivité altérée du réseau par défaut chez des adolescents présentant un Trouble de Stress Post-Traumatique
Le Trouble de Stress Post-Traumatique (TSPT) se caractérise par des symptômes d’intrusions, de reviviscence, d’évitement et d’hypervigilence. Ces symptômes pourraient…

Un lien causal entre une diminution des taux d’acides gras polyinsaturés et des déficits motivationnels
Les pathologies psychiatriques, telles que la schizophrénie, les troubles bipolaires ou la dépression majeure sont classiquement considérées comme différentes d’un…
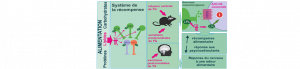
Les lipides nutritionnels contrôlent le système de la récompense
L’alimentation est sans doute essentielle à la survie mais est aussi source de plaisir. Depuis plusieurs années la littérature scientifique…
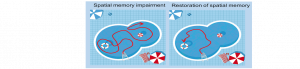
LE METABOLISME DES ASTROCYTES S’INVITE DANS LA MALADIE D’ALZHEIMER
Des chercheurs du Laboratoire des maladies neurodégénératives (CNRS/CEA/Université Paris-Saclay) et du Neurocentre Magendie (Inserm/Université de Bordeaux) viennent de mettre en…

Un engramme ocytocinergique pour apprendre et contrôler sa peur
Nos souvenirs définissent qui nous sommes, qui nous serons. L’idée d’une représentation physique de la mémoire remonte à il y…
SNE Impact 2019
Chaque année, la Société de Neuroendocrinologie sélectionne une dizaine de publications majeures qui reflètent le dynamisme des différents domaines de recherche…