Étiquette : Actualité

La Lettre des Neurosciences
Le numéro 70 de la Lettre vient de paraître. A découvrir en ligne ! Lire la Lettre

Session de NeuroMentoring
Ne manquez pas la prochaine session de NeuroMentoring organisée par le Bureau des Jeunes Chercheur·e·s sur le thème « Impostor Syndrome…
Faits marquants 2025
Les Faits Marquants 2025 sont en ligne ! Pour les consulter : https://www.neurosciences.asso.fr/faits-marquants/ Image : © Wiegering A et…

Coupes budgétaires au CNRS et à l’ESR : un décrochage organisé qu’il faut refuser
La Société des Neurosciences prend position sur les coupes budgétaires imposées au CNRS et à l’ESR. Nous soutenons la mobilisation…

Lecture Alfred Fessard 2026
La lecture Alfred Fessard 2026 sera donnée par Claire Rampon (CRCA, CBI, Toulouse, France) sur le thème : « How adult…

New deadline – Call for symposium proposals
The NeuroFrance 2027 call for symposium is now open! 🤩 Shape this event with us by submitting your proposals before…
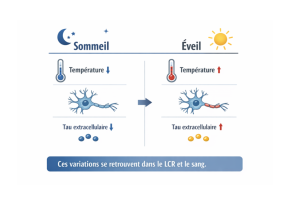
Turn on the Heat ! Le rôle clé de notre température corporelle dans la maladie d’Alzheimer
Notre cerveau fonctionne mieux quand ses grands rythmes physiologiques sont stables, notamment le sommeil et la température corporelle. Dans notre…

Zoom sur nos actions 2025
En 2025, la Société des Neurosciences s’est affirmée comme un acteur engagé du débat public scientifique. Elle a pris position…

Un rythme cérébral du sommeil apparu il y a 300 millions d’années
Un rythme global ultra-lent, cérébral et corporel, spécifique au sommeil profond des mammifères vient d’être également identifié chez sept espèces…

Prix FENS Forum 2026
Doctorant·e·s et post-doctorant·e·s membres de la Société des Neurosciences, candidatez pour un soutien financier pour participer au Forum FENS 2026…