Étiquette : Actualité
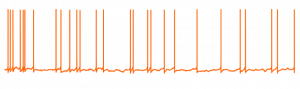
Un nouveau mécanisme pour comprendre l’instabilité des circuits neuronaux dans la maladie d’Alzheimer
Il existe un consensus croissant sur le fait que la maladie d’Alzheimer (MA) implique une défaillance de la machinerie homéostatique,…

Pétition adressée à la Commission Européenne
La Société des Neurosciences exprime son soutien à la lettre ouverte demandant à la Commission Européenne la révision du titre…

Le recyclage cellulaire contre la mort neuronale
Les synucléinopathies sont des maladies neurodégénératives caractérisées par l’accumulation d’une protéine appelée α-synucléine sous forme agrégée au sein des cellules…
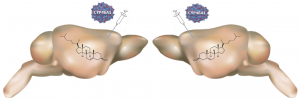
Réguler le métabolisme du cholestérol protège les neurones
Le cholestérol participe à des fonctions essentielles du cerveau, et des dysfonctions de son métabolisme sont observées dans plusieurs maladies…

Loi de programmation pluriannuelle de la recherche : rapports disponibles
Les rapports des trois groupes de travail remis au Premier Ministre sont à présent disponibles sur le site internet…

La Société recrute un(e) Community manager
A l’occasion de la Semaine du Cerveau, la Société des Neurosciences recrute un(e) Community manager. Missions : Animation et gestion…

Le cervelet stabilise notre carte mentale
Savoir s’orienter pour trouver son chemin vers une destination donnée est une fonction essentielle à chaque être vivant. Fermez les…
Loi de programmation pour la recherche : propositions d’un collectif de sociétés savantes académiques
Collectif de Sociétés Savantes Académiques signataires : Société Française de Biologie du Développement, Société Mathématique de France, Société Française…
Concours Beautiful Science – Les lauréats
La Société Française de Physique et ses partenaires, dont la Société des Neurosciences, ont le plaisir de dévoiler les lauréat(e)s…

Activité cérébrale lors de tâches complexes révélée par la neuroimagerie ultrasonore ultrarapide
L’électrophysiologie, plus récemment les mesures optiques mais également les techniques d’imageries magnétiques, ont permis d’enregistrer des évènements associés directement ou…