Tag: Actualité
FENS Forum 2024 Awards
Doctorant·e·s et post-doctorant·e·s membres de la Société des Neurosciences, candidatez pour un soutien financier pour participer au Forum FENS 2024…
New partnership with the Chen Institute
The French Neuroscience Society, whose aim is to promote and support the development of research in all areas of neuroscience…
Participation de mécanismes épigénétiques au déficit mnésique associé à la maladie de Huntington
La maladie de Huntington (MH) est une maladie neurodégénérative héréditaire qui débute le plus souvent à l’âge adulte et entraîne…

Traiter des défauts transitoires précoces retarde les signes de la maladie de Huntington chez la souris
La maladie de Huntington (MH) est due à la mutation du gène huntingtine, un gène qui s’exprime dès les premiers…
Valérie Crépel, lauréate du Prix Innovation Inserm 2022
En moins de dix ans, les neurobiologistes Valérie Crépel (unité 1249 Inserm/Aix-Marseille Université, Marseille) et Christophe Mulle (Institut interdisciplinaire de…
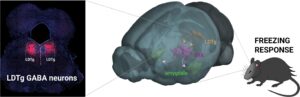
Comment le système de récompense module les réactions de peur?
Le stress est un moteur essentiel de l’adaptation et la réponse au stress d’un organisme est généralement bénéfique car elle…
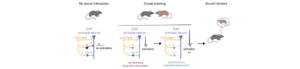
FORMATION DE LA MEMOIRE SOCIALE : PLASTICITES EN CASCADE DANS L’HIPPOCAMPE
La cognition sociale est une fonction importante pour de nombreuses espèces qui est altérée lors de maladies psychiatriques et neurodégénératives.…
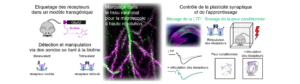
UNE NOUVELLE BOITE A OUTIL POUR EXPLORER LA DYNAMIQUE DES RECEPTEURS DANS LE CERVEAU
Les progrès sur la compréhension du fonctionnement du cerveau sont intrinsèquement liés aux progrès des méthodes d’investigation. La dynamique des…

COMPRENDRE LES PLEURS D’UN BEBE N’EST PAS INNÉ !
Quel parent ne s’est pas demandé ce que disent les pleurs de son bébé ? Alors que l’on fait habituellement confiance…
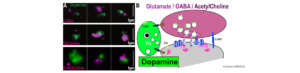
Les Synapses Pivot à Dopamine dans le striatum : un nouveau point névralgique pour la neuromodulation par la dopamine ?
Comment s’organise la conversation entre les neurones dans le cerveau ? Au travers de 2 articles récents nous décrivons une…