Le double jeu de l’APP: fonction et dysfonction neuronale
La maladie d’Alzheimer (MA) est la principale cause de démence. Elle se caractérise par l’association de deux types de lésions: (i) la dégénérescence neurofibrillaires constituées de protéines Tau hyperphosphorylées et (ii) des plaques amyloïdes extracellulaires formées d’agrégats de peptides β-amyloïdes (Aβ) résultant du clivage de la protéine précurseur de l’amyloïde (APP). L’identification de mutations dans les formes autosomiques dominantes à début précoce a placé le métabolisme anormal de l’APP au centre de la maladie, renforçant ainsi l’hypothèse de la cascade amyloïde: la surproduction de peptides Aβ – en particulier les formes plus longues que l’on pense être plus neurotoxiques – pourrait conduire à (ou favoriser) la pathologie Tau et la mort neuronale. Cependant, une grande partie des acteurs moléculaires régulant le metabolisme de l’APP n’a pas encore été caractérisée.
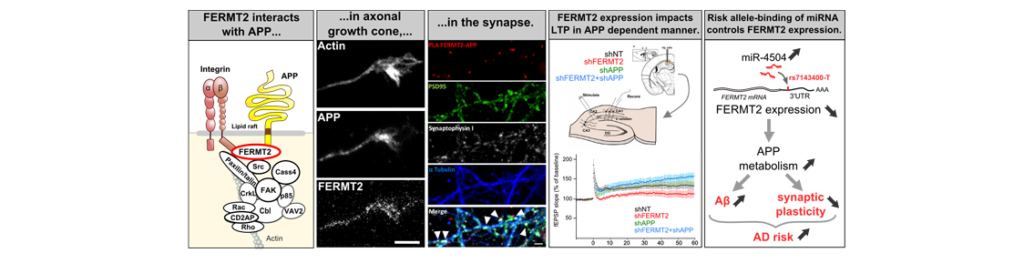
De façon intéressante, les études d’association à l’échelle du génome (GWAS) ont identifié un grand nombre de gènes augmentant le risque de développer les formes les plus courantes de la MA. On peut raisonnablement supposer que certains de ces facteurs génétiques seraient impliqués dans le métabolisme de l’APP et la production d’Aβ. Dans ce contexte, nous avons combiné deux criblages à haut débit à l’échelle du génome pour évaluer l’impact fonctionnel des gènes et microARNR (miRNA) sur le métabolisme de l’APP. Cette approche a mis en évidence l’implication du un facteur de risque génétique FERMT2 (ou Kindlin-2), en tant que modulateur clé potentiel du guidage axonal; un processus neuronal qui dépend de la régulation du métabolisme de l’APP. Nous avons constaté que FERMT2 interagit directement avec APP pour moduler son métabolisme et que la sous-expression de FERMT2 affecte la croissance axonale, la connectivité synaptique et la potentialisation à long terme d’une manière dépendante de l’APP. Enfin, l’allèle rs7143400-T, associé à un risque accru de MA et localisé dans le 3’UTR de FERMT2, a induit une régulation à la baisse de l’expression de FERMT2 par liaison du miR-4504. Ce miARN est principalement exprimé dans les neurones et significativement surexprimé dans les cerveaux de patients par rapport aux témoins. Dans l’ensemble, nos données fournissent des arguments en faveur d’un effet déletère de la sous-expression de FERMT2 dans les neurones et un aperçu de la façon dont cela peut influencer le développement de la MA.
Reference: Alzheimer’s genetic risk factor FERMT2 (Kindlin-2) controls axonal growth and synaptic plasticity in an APP-dependent manner. Eysert F, Coulon A, Boscher E, Vreulx AC, Flaig A, Mendes T, Hughes S, Grenier-Boley B, Hanoulle X, Demiautte F, Bauer C, Marttinen M, Takalo M, Amouyel P, Desai S, Pike I, Hiltunen M, Chécler F, Farinelli M, Delay C, Malmanche N, Hébert SS, Dumont J, Kilinc D, Lambert JC, Chapuis J. Mol Psychiatry. 2020 Nov 3. doi: 10.1038/s41380-020-00926-w. Online ahead of print. PMID: 33144711
Contact chercheur: Julien Chapuis, PhD
Unité INSERM-1167, “Facteurs de risque et déterminants moléculaires des maladies liées au vieillissement”, Lille.
Alzheimer’s disease (AD) is the principal cause of dementia. AD is characterized by two main pathological hallmarks: (i) intracellular neurofibrillary tangles consisting of hyper-phosphorylated Tau proteins and (ii) extracellular amyloid plaques consisting of aggregates of β-amyloid (Aβ) peptides resulting from the processing of amyloid precursor protein (APP). The identification of early-onset autosomal dominant AD-linked mutations have placed abnormal APP metabolism at the center of the disease, further supporting the amyloid cascade hypothesis: the overproduction of Aβ peptides –especially the longer forms that are thought to be more neurotoxic– may lead to (or favor) Tau pathology and subsequent neuronal death. However, a large proportion of the key molecular players in APP trafficking have yet to been characterized.
Interestingly, Genome-Wide Association Studies (GWASs) identify a large number of novel risk-increasing loci in common late-onset form of AD. One can reasonably assume that some of these genetic factors are involved in APP metabolism and Aβ production. In this context, we combined two genome-wide high-content screenings to assess the functional impact of miRNAs and genes on APP metabolism. This approach highlighted the involvement of FERMT2 (or Kindlin-2), a genetic risk factor of AD, as a potential key modulator of axon guidance; a neuronal process that depends on the regulation of APP metabolism. We found that FERMT2 directly interacts with APP to modulate its metabolism and that FERMT2 under-expression impacts axonal growth, synaptic connectivity and long-term potentiation in an APP-dependent manner. Lastly, the rs7143400-T allele, which is associated with an increased AD risk and localized within the 3’UTR of FERMT2, induced a down-regulation of FERMT2 expression through binding of miR-4504 among others. This miRNA is mainly expressed in neurons and significantly overexpressed in AD brains compared to controls. Altogether, our data provide strong evidence for a detrimental effect of FERMT2 under-expression in neurons and insight on how this may influence AD pathogenesis.